Simulation de Matériaux
Mon équipe de recherche développe des matériaux innovants pour soutenir les technologies à l’hydrogène. Ceci inclut les catalystes moléculaires ou solides pour la production et la conversion énergétique de l’hydrogène, les hydrures métalliques pour le stockage de l’hydrogène, et les matériaux d’électrode pour le stockage de l’électricité dans des batteries ou des supercondensateurs.
Calculs Haute-précision de Structures Électroniques
Le calcul ab initio (à partir de principes premiers) consiste en la simulation des atomes et des électrons qui composent un matériau, et la résolution des équations de la mécanique quantique pour en prédire toutes les propriétés physiques. Ces calculs se basent sur la théorie de la fonctionnelle de densité (DFT), qui permet de calculer l’énergie d’un système dans son état fondamental ainsi que la fonction d’onde des électrons. Nous utilisons la DFT pour prédire des paramètres structurels d’une molécule ou d’un matériau, ainsi que l’énergie associée à une réaction chimique ou à la formation d’un matériau.
D’autres techniques plus avancées permettent de mieux décrire les niveaux énergétiques des électrons ou la réponse d’un matériau à une perturbation externe. Par exemple, la méthode GW permet de prédire l’énergie nécessaire pour ajouter ou retirer un électron d’un système, et la méthode BSE permet de calculer les propriétés optiques des matériaux.
Gabriel Antonius est physicien spécialisé en simulations numériques des matériaux. Il a complété son doctorat en physique à l’Université de Montréal, puis a travaillé durant trois ans comme chercheur postdoctoral à University of California Berkeley, et au Lawrence-Berkeley National Laboratory. Il est professeur à l’Université du Québec à Trois-Rivières depuis 2018.
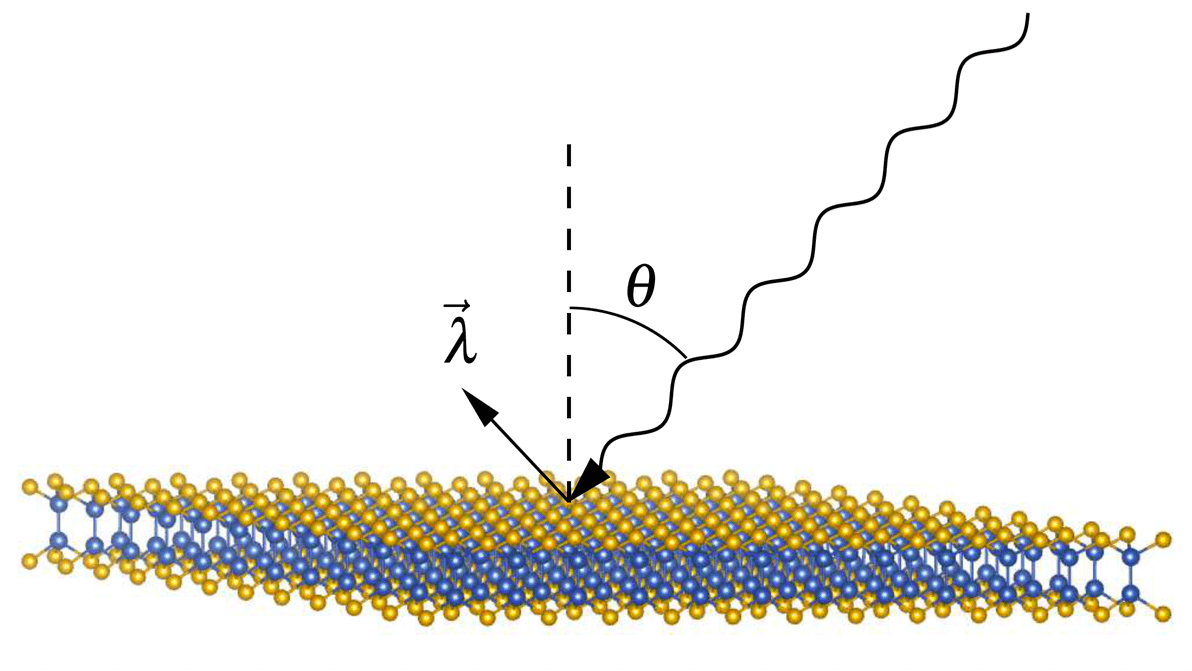
Matériaux Énergétiques pour un Futur Durable
La production et le stockage des énergies renouvelables constitue un des plus grands défis à relever pour faire face à la crise climatique. L’hydrogène est un vecteur énergétique particulièrement adéquat pour les énergies vertes. Le processus d’électrolyse convertit la puissance électrique en énergie chimique en séparant les molécules d’eau en oxygène et en hydrogène gazeux. L’hydrogène est ensuite convertis en électricité dans une pile à combustible, et peut ainsi propulser une voiture électrique.
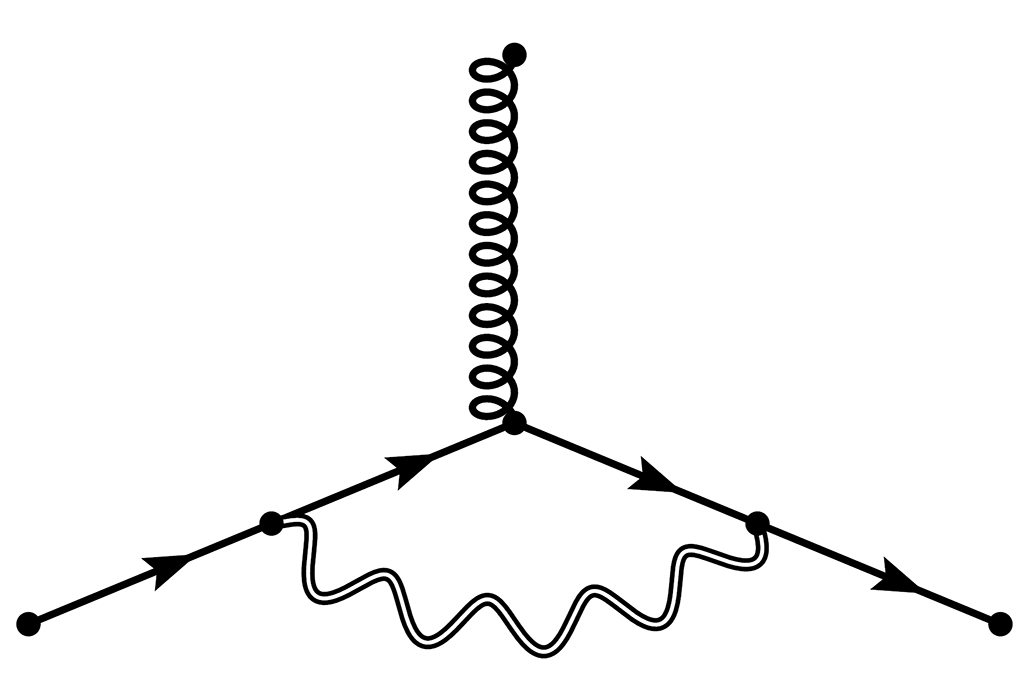
Couplage Électron-Phonon
Les phonons décrivent les modes de vibrations des atomes dans un solide, et sont en quelque sorte des particules de son, ou des quanta de vibrations. Lorsque les électrons se déplacent dans un solide, ils subissent des collisions avec les phonons et ce processus est au coeur de multiples phénomènes, tels que la résistivité électrique, la supraconductivité, et le changement des propriétés optiques en fonction de la température. Nous étudions ces phénomènes par calcul ab initio à l’aide de la théorie de la fonctionnelle de densité perturbative (DFPT).